
To play this video, please allow cookies and other external services from YouTube.


To play this video, please allow cookies and other external services from YouTube.
To play this video, please allow cookies and other external services from YouTube.
Sealing devices enable the contamination-free storage of sterilized instruments until they are needed for the patient treatment. We provide routine tests such as the “seal seam stability test” or MELAink test to check the reliability and safety of the packaging seal seam. These tests should be performed at intervals established by regional authorities.
The hospital sector has long used sealing devices which perform an automatic check during the sealing procedure, documenting the three process parameters temperature, contact pressure and sealing time. The use of such validatable sealing devices in practice-based medicine is currently not mandatory in many countries, but often recommended.
Nevertheless, practices and clinics considering a new purchase should consider to choose a validatable sealing device, guaranteeing reproducible safety through continual monitoring and documentation of all the process parameters.
The MELAG sealing devices MELAseal Pro and MELAseal 200 are fitted with a process-parameter monitoring system as prescribed by international standards. Sealing logs are issued via the documentation interfaces.
The requirements placed on a medical device are set out in the directive 93/42/EEC. In accordance with article 1 of the directive, a product is a medical device if section 2 of 93/42/EEC is applicable.
Neither section 2a, which describes products which are applied in or on the human body, nor section 2b (medical devices accessories) contains a description of the sealing device and its application. Appendix IX of the classification criteria does not provide a definition of a sealing
device as a medical device.
As such, sealing devices are not classed as a medical device. Nevertheless, in developing and producing its sealing devices, MELAG orients itself around the requirements of directive 93/42/EEC.
The transport, commissioning and storage of sterile medical devices including the requisite packaging and marking are regulated by international standards. Loss of sterility is dependent less on the length of the storage time as from external influences during storage,
transport and application. It is not possible to establish a storage duration for practice and clinic use.
The recommendation pertaining to the storage duration of medical devices is: paper and transparent packaging or another equivalent packaging can be stored in protected storage (in drawers, cupboard etc.) for six months. The storage duration of the instruments may not exceed the individually specified expiry date.
Packaging systems (combination of a sterile barrier system and protective packaging) can be stored for 5 years, as far as the manufacturer has not determined an alternative expiry date.
Unwrapped instruments should be used within 24 and 48 hours. Please always consider your national requirements.
The rooms used for storing medical devices should be dry, dark, cool and easy to clean. They must not be accessible to everyday activity. We recommend protected storage of the instruments in rooms and cupboards which comply with the requirements of international standards.
MELAfol is a transparent sterilization packaging (available as a roll or various size bags) perfectly adapted to MELAG sealing devices and complying with international standards. MELAfol is tear-free, sealed against germs and easy to open. Find out more about our MELAfol products for safe instrument storage after sterilization and approval.
In instrument reprocessing, a sealing device is used to seal sterilisation pouches or films securely and with absolute reproducibility so that the previously sterilised instruments packaged in them remain reliably barrier-protected until they are used. After cleaning, disinfection and sterilisation, the instruments are placed in suitable sterilisation packaging, which is then sealed in the sealing device using defined parameters such as temperature, sealing time and contact pressure. This controlled process ensures that each seal seam is consistently tight and stable and fulfils the normative requirements. Only a correctly executed and documented seal can prevent germs or other contaminants from entering the packaging from the outside and jeopardising the sterile condition. The sealing device therefore makes a significant contribution to maintaining the sterility of the instruments during storage, transport and provision, thus ensuring safe use on patients.
There are two main types of sealing devices in instrument reprocessing, which differ in their mode of operation and scope of performance. Continuous flow sealers work continuously: the sterilisation pouches or tubes are simply fed into the feed opening, transported automatically and sealed at a constant speed. As a result, they achieve a particularly high throughput and are ideal for practices or clinics with a high volume of instruments.
Impulse sealers, on the other hand, work in cycle mode. Here, the pouch is inserted manually and sealing takes place after the sealing arm is closed or by activating the sealing plate. These devices are more compact, require less space and are suitable for small to medium quantities of sealed goods.
Both types of device can be validated, which is crucial in the medical sector. Regardless of the design, the sealing device must guarantee consistent and reproducible process quality - this is the only way to ensure that each seal fulfils the requirements for stability, tightness and conformity to standards.
The use of sealing devices in instrument reprocessing is subject to clear standards and specifications that define the requirements for both the packaging material and the sealing process itself. DIN EN ISO 11607-1 and DIN EN ISO 11607-2 are central to this.
DIN EN ISO 11607-1 describes the basic requirements for sterile barrier systems and their packaging materials. It specifies the properties that sterilisation packaging must have in order to ensure the sterile status of the goods during sterilisation, storage, transport and use.
DIN EN ISO 11607-2 regulates the requirements for process validation of the sealing procedure. It requires that the sealing process is demonstrably controlled and reproducible - i.e. works with constant parameters such as temperature, pressure and time. This ensures that every sealed seam is reliable and stable.
In addition, material-related standards apply, in particular the EN 868 series, which describes further specific requirements for sterilisation packaging, e.g. with regard to strength, puncture resistance or material structure.
In addition to the standards, the manufacturer's specifications for the sealing devices and packaging materials are also binding. They specify the conditions under which the respective product can be operated in compliance with the standard and which parameters must be adhered to in order to achieve a valid and reliable sealing result.
A validatable sealing process means that the three decisive process parameters of temperature, pressure and time (TDZ) are continuously monitored, recorded and evaluated in order to be able to prove the consistently reliable quality of the sealed seams. Such a process fulfils the requirements of DIN EN ISO 11607-2 and makes it possible to prove at any time that each individual seal has taken place under controlled and stable conditions.
This also includes validation as part of IQ/OQ/PQ:
IQ (Installation Qualification) checks whether the sealing device has been installed correctly and fulfils all technical requirements.
OQ (Operational Qualification) ensures that the device functions reliably within defined parameters, i.e. that it reaches and maintains the target values for temperature, pressure and time.
PQ (Performance Qualification) proves that the entire sealing process produces reproducibly reliable sealing seams in the actual workflow.
This monitoring and documentation makes it possible to prove at any time that the sealing seams fulfil the normative requirements and thus guarantee verifiably safe, stable and leak-proof packaging of the sterile goods.
Typical sealing parameters always depend on the packaging material used and the respective sealing device, as different film and paper qualities place different demands on temperature, pressure and time. As a general rule, the parameters must be set in such a way that sufficient seal strength is achieved without causing thermal or mechanical damage to the material.
In practice, the temperatures of many medical sealers are often in the range of approx. 170-190 °C, while the pressure and sealing time vary depending on the type of device. Rotary sealers operate at a constant temperature and transport speed, while impulse sealers regulate the sealing time via a defined sealing time.
The exact settings are not specified across the board, but are defined and documented during validation (IQ/OQ/PQ). This involves checking which combination of parameters produces a stable, standard-compliant and reproducible sealed seam for the material used. The decisive factor is therefore not a specific numerical value, but process consistency and the verifiable suitability of the parameters for the respective packaging system.
Only approved and standard-compliant packaging materials that have been specially developed for steam sterilisation are suitable for sterile packaging in instrument reprocessing. These include, in particular, sterilisation films and papers, transparent pouches and sterilisation tubes or rolls that meet the requirements of DIN EN ISO 11607 and the EN 868 series.
These materials consist of suitable film and paper composites that ensure the necessary barrier effect against germs on the one hand and allow steam to enter and exit safely during sterilisation on the other. Only tested and approved original materials reliably fulfil these requirements.
It is also important to only use original packaging with batch labelling. Batch labelling enables clear traceability and is a prerequisite for correct documentation as part of quality assurance. Material batches that are clearly tested and certified ensure that each packaging has the required stability, tear resistance and sterile barrier performance.
To summarise: Standard-compliant sterilisation films, papers, pouches and rolls that are specifically intended for medical sterilisation are suitable - and may only be used in original quality with traceable batch labelling.
Testing the seal seam quality on a daily basis involves several practical and standardised inspection steps to ensure that each pack forms a stable and tight sterile barrier. In routine operation, this begins with the visual inspection of each individual seal seam: the seam must be even, continuous and pore-free, without interruptions, creases or burnt areas. Deviations such as shadows, fraying or irregular structures can be indications of faulty sealing parameters or material problems.
Peel tests are also carried out regularly as part of the inspection plan. The seal seam is torn open in a controlled manner to assess whether the film and paper separate cleanly without the material tearing or fibres being torn out. The peel test shows whether the joint is stable enough but can still be opened in a controlled manner, as required for use in sterile areas.
Regular strength tests (e.g. with standardised test strips or tensile testers) are also part of quality assurance. These tests ensure that the seal seams achieve the required strength and that the packaging does not tear open unintentionally during storage, transport and handling.
All test results must be documented in accordance with the in-house test plan. Only complete documentation provides proof of a consistently reliable sealing process and fulfils the requirements of the applicable standards and guidelines.
Correct labelling of sterile packaging is essential for traceability, documentation and patient safety. It includes several mandatory details that must be applied clearly, legibly and abrasion-resistant. This usually includes
Sterilisation date and expiry dateThe sterilisation date documents the time of reprocessing. The expiry or release date is determined by the in-house storage periods for packaged sterile goods.
Batch and device IDEach reprocessing unit requires a unique batch identification that can be used to trace the sterilisation cycle, the device used and any employees involved. The device ID or steriliser name must also be included.
UDI/barcode (if required)For certain medical devices or internal QM processes, a UDI (Unique Device Identification) or a machine-readable barcode may also be required to enable seamless digital traceability.
Ideally, labelling is carried out directly using the integrated print function of modern validatable sealing devices, which automatically print the information cleanly and smudge-proof onto the packaging. Alternatively, external label printers or pre-printed, abrasion-resistant labels can be used. It is important that the labelling remains permanent - even during storage, transport and handling - and does not cause any damage to the sterile barrier.
The integration of a sealing device into traceability is an important part of a modern, documentation-secure reprocessing process. Today, validatable sealing devices usually have digital interfaces such as USB or LAN, which can be used to automatically transfer all relevant process parameters and batch data to a documentation or reprocessing management system (DMS/AEMP software). As a result, temperature, pressure, time as well as device and operating information are seamlessly archived and linked to the respective sterile goods.
In addition, barcode scanners or integrated print and scan modules can be used to clearly link individual pouches to the corresponding sterilisation batch. This creates a direct link between the sealing process, sterilisation cycle and subsequent use on the patient or case. When sealing, for example, the employee can scan the pouch, the steriliser batch and - depending on the workflow - even the patient case so that all data records are automatically linked.
In this way, the sealing device becomes an integral part of digital traceability: every step - from packaging and sealing to sterilisation and subsequent use - is clearly traceable. This not only increases patient safety, but also fulfils the requirements for audit-proof, standard-compliant documentation.
Common sealing defects are usually caused by incorrect process parameters, unsuitable handling or contaminated packaging material. Typical problems are
UndersealingThe seal seam is not fully bonded, appears porous or is too easy to open. This is usually caused by too low a temperature, too short a sealing time or insufficient contact pressure. Prevention: Check sealing parameters, comply with validation values, maintain the appliance regularly.
Over-sealingThe seam is burnt, too wide or shows material damage. This is often caused by too high a temperature or too long sealing contact. Prevention: Adjust the temperature and sealing time to the material requirements, follow the manufacturer's instructions.
"Shadow seams"Irregular, patchy or inhomogeneous seam structures that indicate inadequate heat or pressure distribution. Avoidance: Check the contact pressure, adjust the transport speed (for rotary sealers), operate the device rhythmically and evenly.
Wrinkles or creases in the materialWrinkles can result in the seam not closing completely and the sterile barrier being interrupted. Avoidance: Insert pouches cleanly and smoothly, select the correct cutting length for roll material, check material tension.
Contaminated or moist sealing surfacesResidues such as moisture, powder, instrument oil or cleaning agents prevent a stable bond. Avoidance: Only use completely dry packaging, keep the working environment clean, do not bring films and paper into contact with wet hands or contaminated surfaces.
The width of the sealing seam is an important quality parameter because it contributes significantly to the stability and tightness of the sterile barrier. In practical use, the sealing seam should be at least around 6 mm wide - this value is a recognised minimum standard, but can vary depending on the device type, material and manufacturer specifications. Many modern sealing devices even produce wider seams to create additional safety reserves.
The actual seam width required is determined during validation. This involves testing and documenting which seam width provides sufficient strength and a standard-compliant barrier effect for the packaging material used. The decisive factor is therefore not just a general millimetre value, but that the seam width achieved is constant, reproducible and demonstrably suitable.
The shelf life of sterile goods cannot be specified with a fixed date, as it depends on several factors. The packaging material, the storage conditions and the handling of the packaged sterile goods are the most important factors. The basic rule is: as long as the packaging is undamaged, dry, clean and intact, the sterile condition is maintained - however, practical storage periods are set to ensure safety and traceability.
Clean, dry and low-dust storage contributes significantly to extending the shelf life. Closed cabinets or shelves in an area with as little air turbulence as possible and without moisture ingress are ideal. Careful handling is equally important: kinks, crushing or abrasion on the packaging can jeopardise the sterile barrier and significantly reduce the shelf life.
The first-in, first-out (FIFO) principle is recommended for organisational safeguarding. This ensures that older batches are used first and that no packaging is stored for an excessively long time.
A sealing device requires regular and professional maintenance to ensure permanently reliable, reproducible sealing seams. This primarily includes cleaning the pressure rollers and heating elements, as particles, fibres or film residues can build up there in everyday use and impair the seam quality. Clean contact surfaces ensure even heat and pressure transfer.
It is equally important to check and replace wearing parts in good time - for example rollers, Teflon tapes, heating strips or transport components. Worn or damaged components can lead to shadow seams, irregular structures or incorrect sealing.
Calibrating the temperature sensors is also a key part of maintenance. A reliable and validatable sealed seam can only be produced if the device measures and controls the actual temperature correctly. Deviations in temperature control often only become apparent during tests or in the validation process, which is why regular calibration is absolutely essential.
All measures - whether cleaning, replacing parts, functional tests or calibrations - must be recorded and documented as part of regular validation. This provides proof that the device is always operated in compliance with standards and fulfils the requirements for a reliable, reproducible sealing process.
Selecting the right sealing device for a practice or clinic means taking into account both the technical requirements and the organisational framework conditions for reprocessing. A decisive factor is performance: practices with a large volume of instruments often benefit from rotary sealers, which enable a fast and continuous workflow. Impulse sealers can be perfectly suitable for low to medium volumes.
Material mixing is also important. If bags of different sizes or rolls are frequently processed, the sealer must be flexible and ergonomic to operate. The degree of integration also plays an important role: modern devices with integrated print function, barcode support and computer connection (USB/LAN) facilitate traceability and reduce manual work steps. Whoever uses digital documentation must ensure a seamless connection to the reprocessing software.
The space requirements and the spatial structure of the reprocessing unit also influence the selection. Compact pulse devices fit well in small sterile rooms, while continuous flow devices usually require more space and optimised work logistics.
Another point is the validation requirements. The device must guarantee a stable and reproducible process and be able to document all necessary parameters. The higher the requirements of the practice or clinic - e.g. in relation to audits or quality management systems - the more important it is to choose a validatable device with precise process control.
The total cost of ownership (TCO ) should also be taken into account. This includes not only the purchase price, but also ongoing costs such as maintenance, consumables, service contracts and possible downtime. Good service from the manufacturer and the availability of regional technicians are essential for trouble-free operation.
Requalification refers to the re-examination of an already validated sealing process to ensure that it continues to work stably, reproducibly and within the defined tolerances. It is an integral part of quality assurance in accordance with DIN EN ISO 11607-2 and is part of every professional reprocessing process.
Requalification is always due when general conditions change that could have an impact on the sealing process. Typical triggers are
Material changesNew pouch or film materials, other manufacturers or batch changes that involve relevant differences in the material structure.
Parameter adjustmentsIf temperature, pressure or sealing time need to be changed, e.g. due to material changes or optimisations in the workflow.
Change of locationMoving the device - even within the practice - can have an impact on process stability due to temperature, humidity or mechanical influences.
Repairs or technical interventionsReplacement of heating elements, pressure rollers, sensors or electronics requires a mandatory check of the process capability.
Periodic requalification according to the QM planRegardless of changes, requalification is carried out at fixed intervals (e.g. annually) to prove that the process remains stable over the entire utilisation period.
The aim of requalification is always the same: to prove that the sealing process continues to be standard-compliant, validated and safe, so that every sealed seam reliably fulfils the requirements for strength and tightness.
Perform and check a test seal seam once a day before starting work by performing a visual and mechanical check. To perform the peel test, sterilize the sealed film packaging in an autoclave. After removing it from the steam sterilizer, pull apart the seal seams slowly in the peel direction. The visual check is performed to check that the film has completely released itself from the paper. No fraying of more than 10 mm is permitted when opening the film packaging.
Document the results of the peel test and all further routine checks in a verifiable fashion. We provide a MELAG template checklist for this purpose.
The daily Seal Check of the seal seam can be performed with the MELAG Seal Check. MELAG Seal Check is a secure and efficient method with which to verify the seal seam. The contrast on the print sample permits the practice team to determine quickly and easily whether the film layer has fused with the paper side correctly. The Seal Check is inserted in the sterilization pouch and is successful if the complete seal seam contains even discolouration and sharp contours. For the traceable documentation of the routine tests of a sealing device, we offer the MELAconnect App. The app helps you to archive the Seal Checks easily via the camera on your smartphone or tablet.
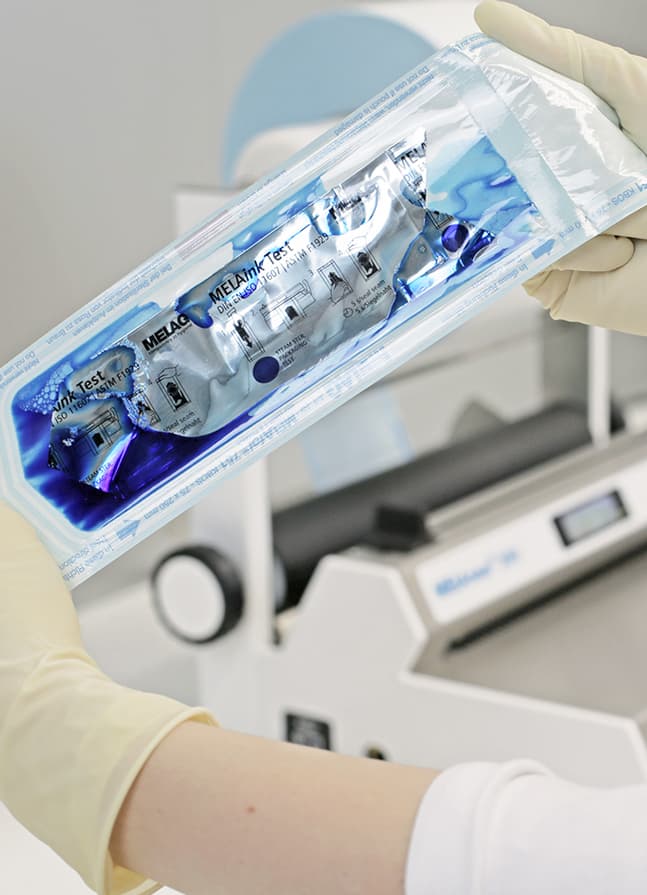
The weekly MELAink Test enables even better recognition of possible irregularities in the seal seam. The ink test is placed in a transparent sterilization package and sealed. Then the ink is pressed out of the test bag in the direction of the seal seams. The spread of the ink tests all four sides of the packaging, revealing irregularities, flaws or channels in the seal seams.
The seal seam stability test is used to perform an annual check of the seal seam for tensile strength (tear-resistance) and validation of the packaging process using MELAG sealing devices. The seal seam stability test is a standardized test procedure for the performance qualification of your sealing device in accordance with international standards. The test is performed with a high-tech electronic test machine for guaranteed valid results. Download the application form for a MELAG seal seam stability test. The form depicts all the necessary steps for performing the seal seam stability test.









